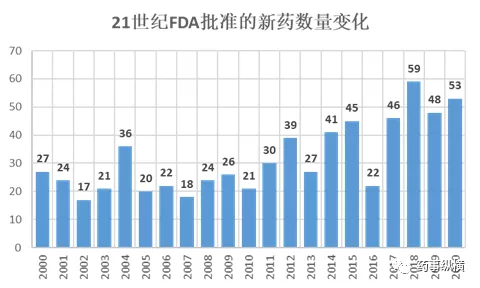
截止2020年12月27日,FDA已经批准了53个新药,其中38个小分子化药,两个RNA,13个生物药。笔者在此对这53个药物进行一一简要介绍,以助业内同仁获得初步性了解。本文中的销售额预测数据均来自Evaluate Pharma网站上公开的文章,因篇幅太长,参考文献和信息来源将不再呈现,但本文经过校对,每一个数据来源都真实可靠,有参考文献可循,如有疑问,可联系voyager88。另外在此声明,版权所有,严禁转载,抄袭必究。
1. Ayvakit(avapritinib)
1月9日,FDA批准了Blueprint Medicines公司的avapritinib,用于阿尔法血小板衍生生长因子受体(PDGFRA)基因外显子18突变阳性,且无法手术切除或转移性的成人胃肠间质瘤患者治疗。胃肠间质瘤是一种比较罕见的肿瘤,美国每年新发病例为5000-6000人[i],其中大约有10%左右的病例携带PDGFRA基因突变。本品是首个PDGFRA基因突变的胃肠间质瘤的针对性疗法,FDA批准本品上市是基于一项有43名患者参与的临床研究(NCT02508532)结果。入组的43名患者全部为外显子18突变,其中38名患者为PDGFRA D842V突变。在接受本品治疗后,总缓解(ORR)率为84%,其中完全缓解(CR)率为7%,部分缓解(PR)率为77%,61%的患者持续缓解时间超过了6个月,而PDGFRA D842V突变的患者亚组中,总缓解率高达89%,其中完全缓解率为8%,其中部分缓解率为82%,59%的患者持续缓解时间超过了6个月[ii]。与治疗相关的3-4级不良反应发生率为57%,最常见的不良反应为贫血(17%)[iii]。市场潜力方面,EvaluatePharma(EVP)预测该产品在2026年的销售额为9.18亿美元。
2. Tepezza(teprotumumab-trbw)1月21日,FDA批准了Horizon Therapeutics的teprotumumab,用于甲状腺眼病治疗。甲状腺眼病又称Graves眼病,是一种自免疫相关的疾病,年发病率为19/10万[iv],该疾病在美国患病相对较低,根据美国罕见疾病组织(NORD)数据,女性患病率为16/10万,男性患病率为2.9/10万[v]。FDA批准teprotumumab是基于两项、共有170名患者参与的临床试验的结果,其中teprotumumab治疗组分别有71%(试验一,NCT01868997)和83%(试验二,NCT03298867)的患者眼球突出高度下降了2mm以上,而安慰剂组患者则分别仅为20%和10%[vi,vii]。Teprotumumab是首个获批用于甲状腺眼病的药物特效药物,市场潜力巨大,根据该公司三季度财报,该产品在过去两个季度的销售额就已达4.76亿美元,并且预测2020年的年销售额将超过8亿美元[viii],而EVP仅预测该产品在2024年的为4.84亿美元。 3. Tazverik(tazemetostat)1月23日,FDA加速批准了Epizyme 公司的tazemetostat,用于转移性、局部转移性或无法手术根除的晚期上皮样肉瘤治疗。上皮样肉瘤是一种非常罕见软组织肿瘤,不足软组织肉瘤的百分之一,据估算美国仅有数百名患者。Tazemetostat是一种甲基转移酶抑制剂,可阻断EZH2甲基转移酶的活性,进而阻止癌细胞的生长,是首个获批用于该疾病的特效药物。FDA加速批准本品上市,是基于一项有62名转移性或局部晚期上皮样肉瘤患者参与的、开放标签的临床研究(NCT02601950)结果。经过本品每日两次800mg治疗后,总缓解率为15%,中位无进展生存期(PFS)为5.5个月,中位总生存期(OS)为19.0个月,获缓解的9名患者中,6名患者的持续缓解时间超过了6个月[ix]。除上皮样肉瘤,tazemetostat还在开发滤泡性淋巴瘤等适应症。在一项2期临床试验中,45名EZH2基因突变型和54名EZH2基因野生型滤泡淋巴瘤患者,分别接受本品治疗后表现出的总缓解率分别为69%和35%,中位持续缓解时间(DOR)分别为10.9个月和13.0个月,中位PFS分别为13.8个月和11.1个月[x]。市场潜力方面,EVP预测该产品在2024年的销售额为5.56亿美元。
4. Pizensy(乳糖醇)2月12日,FDA批准了Braintree公司的乳糖醇,用于成人慢性特发性便秘(CIC)治疗。乳糖醇是一种渗透型泻剂,FDA批准本品的基础是两项临床试验的结果。在其中一项随机、双盲的临床试验(NCT02819297)中,患者分别接受乳糖醇和安慰剂治疗6个月,以每周自发排便数超过3次、12周内至少有9周且最后4周至少有3周的自发排便数相比基线至少增加1次为综合指标,计算应答率。其结果显示,乳糖醇(n=291名)治疗组的应答率为25%,而安慰剂组(n=303)的应答率仅为13%,达预设的主要终点。而另外一项临床试验(NCT02481947)是以鲁比前列酮为对照,评估方法与前一项试验一致,试验结果也相似[xi],但非劣性结果未被FDA采纳。乳糖醇与乳果糖一样,是经典的“泻”药,最早于1993年就获得西班牙批准上市,除了便秘,西班牙还批准乳糖醇用于肝性脑病治疗。
5. Nexletol (bempedoic acid)2月21日,FDA批准了Esperion Therap公司的bempedoic acid,用于在饮食控制和他汀类最大耐受剂量治疗的下,仍需要进一步降低脂蛋白(LDL-C)的杂合子高胆固醇血症或动脉粥样硬化的心血管疾病患者治疗。Bempedoic acid是一种新型口服降脂药,可通过抑制三磷酸腺苷-柠檬酸裂解酶(ACL)的活性,降低肝脏对LDL-C的合成。FDA批准本品上市是基于两项随机、双盲的临床研究结果。在第一项临床试验(NCT02666664)中,2330名患者在接受最大耐受剂量的降脂疗法(最大剂量的他汀或他汀类联用其它药物,但不含使用40mg及以上辛伐他汀或PCSK9抑制剂的患者)的基础上,按2:1的比例分别添加本品或安慰剂治疗,结果显示本品治疗组在第12周的LDL-C水平相比基线下降了17%,而安慰剂组仅为2%,达到统计学显著性差异[xii]。另一项临床试验(NCT02991118)的设计和结果与前一项试验相似,bempedoic acid和安慰剂治疗组在第12周的LDL-C水平分别相比基线下降15%和2%[xiii]。市场潜力方面,虽然本品面临着PCSK9抑制剂的竞争,但美国有超过50%的患者对他汀类不耐受或单独使用他汀类无法有效控制血脂,EVP预测本品在2024年的销售额为7.16亿美元。
6. Vyepti(eptinezumab-jjmr)2月21日,FDA还批准了灵北的降钙素基因相关肽(CGRP)单抗eptinezumab上市,用于预防性治疗偏头痛。本品的获批是基于两项临床研究的结果,在试验一(NCT02559895)中,665名阵发性偏头痛患者按1:1:1的比例,分别每三月一次接受安慰剂、本品100mg或300mg治疗,持续治疗12个月,主要终点为1-3个月内平均每月偏头痛发作天数相比基线值的变化。结果显示,安慰剂、本品100mg或300mg治疗组患者的月均偏头痛发作天数分别相比基线下降3.2天、3.9天和4.2天,eptinezumab的两个剂量组均与安慰剂组达到统计学显著性差异。在另一项针对慢性偏头痛的临床试验(NCT02974153)中,共计有1072名患者入组,试验的设计与试验一类似,试验结果显示,安慰剂、本品100mg或300mg治疗组患者的月均偏头痛发作天数分别相比基线下降5.6天、7.7天和8.2天,eptinezumab的两个剂量组也均与安慰剂组达到统计学显著性差异[xiv]。市场方面,CGRP靶点竞争较为激烈,此前FDA已经批准了安进诺华的erenumab、梯瓦的fremanezumab、礼来的galcanezumab和Biohaven的rimegepant,EVP预测本品在2024年的销售额仅为4.04亿美元。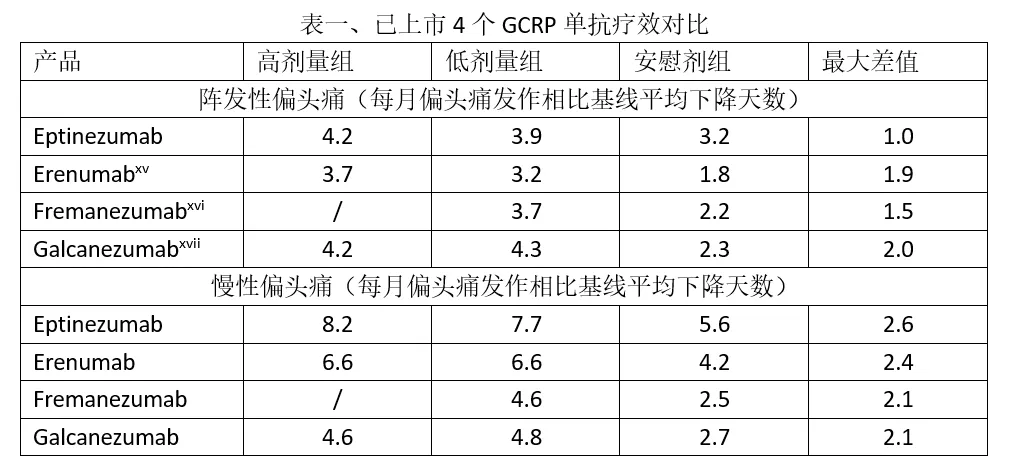 7. Barhemsys (氨磺必利)2月26日,FDA批准了Acacia公司的氨磺必利注射液,用于术后呕吐预防和治疗。氨磺必利是一个多巴胺D2和D3受体阻断剂,上世纪80年代开始应用于精神分裂症治疗。FDA批准氨磺必利用于术后呕吐治疗和预防的依据是4项临床研究的结果,其中试验一和实验二是以预防为设计目的的临床试验,而试验三和试验四则是以治疗为设计目的。试验一(NCT01991860)[xviii]的患者均接受本品或安慰剂单独预防(n=342),试验二(NCT02337062)[xix]的患者均接受本品或安慰剂联合其它机制的止吐药共同预防(n=1147),试验三(NCT02449291)[xx]的患者均为初治患者(n=369),试验四((NCT02646566)[xxi]的患者均为经治患者(n=465),临床终点均为完全缓解(试验一、二定义为术后24小时内无呕吐发作或不使用急救药物,试验三、四定义为使用药物治疗后24小时内无呕吐发作或不使用急救药物)率,结果显示,4项临床试验的完全缓解率依次分别为(治疗组 vs 安慰剂组):78% vs 54%、58% vs47%,31% vs 22%和42% vs 29%,均达到统计学显著性差异。市场潜力方面,EVP预测本品在2024年的销售额为1.03亿美元。
7. Barhemsys (氨磺必利)2月26日,FDA批准了Acacia公司的氨磺必利注射液,用于术后呕吐预防和治疗。氨磺必利是一个多巴胺D2和D3受体阻断剂,上世纪80年代开始应用于精神分裂症治疗。FDA批准氨磺必利用于术后呕吐治疗和预防的依据是4项临床研究的结果,其中试验一和实验二是以预防为设计目的的临床试验,而试验三和试验四则是以治疗为设计目的。试验一(NCT01991860)[xviii]的患者均接受本品或安慰剂单独预防(n=342),试验二(NCT02337062)[xix]的患者均接受本品或安慰剂联合其它机制的止吐药共同预防(n=1147),试验三(NCT02449291)[xx]的患者均为初治患者(n=369),试验四((NCT02646566)[xxi]的患者均为经治患者(n=465),临床终点均为完全缓解(试验一、二定义为术后24小时内无呕吐发作或不使用急救药物,试验三、四定义为使用药物治疗后24小时内无呕吐发作或不使用急救药物)率,结果显示,4项临床试验的完全缓解率依次分别为(治疗组 vs 安慰剂组):78% vs 54%、58% vs47%,31% vs 22%和42% vs 29%,均达到统计学显著性差异。市场潜力方面,EVP预测本品在2024年的销售额为1.03亿美元。
8. Nurtec ODT(rimegepant)2月27日,FDA批准了Biohaven公司的口服CGRP抑制剂rimegepant,用于成人偏头痛的急性治疗。Rimegepant不同于CGRP单抗,该产品的定位是快速止痛,为了加快药物的起效速度,Biohaven还将其制成了口崩片。FDA批准本品上市是基于一项随机、双盲的临床研究((NCT03461757)的结果,受试者分别接受本品75mg(n=732)和安慰剂(n=734)治疗,主要终点为单次服药后2小时内无痛患者应答率和无自主指定的最明显症状(MBS,主要是畏光、恶心、恐声)患者应答率。结果显示,本品治疗组的无痛应答率为21.2%,无最明显症状应答率为35.1%,而安慰剂组分别仅为10.9%和26.8%,均达到统计学显著性差异[xxii]。另一项相似设计、有1186名患者参与的多中心临床试验(NCT03237845)也证实了本品的有效性,本品治疗组无痛应答率为19.6%,无最明显症状应答率为37.6%,而安慰剂组分别仅为12.0%和25.2%[xxiii]。市场潜力方面,因为是第一个口服CGRP抑制剂,而且市场定位也与CGRP单抗不同,EVP预测该产品在2024年的销售额可达8.68亿美元。
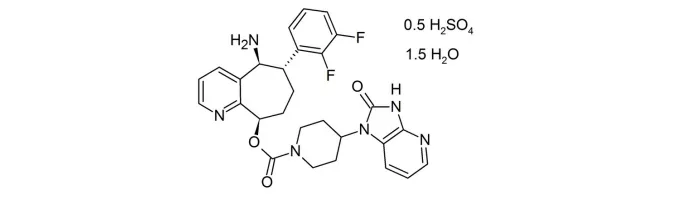
9. Sarclisa(isatuximab-irfc)3月2日,FDA批准了赛诺菲的isatuximab,适应症为联合泊马度胺和地塞米松用于既往接受过两种以上疗法(包括泊马度胺和蛋白酶体抑制剂)的多发性骨髓瘤(MM)治疗。Isatuximab 是一种Anti-CD38抗体,通过抑制其ADP核糖环化酶(ADP-ribosyl cyclase)活性而发挥生理作用。一项名为ICARIA-MM (NCT02990338)的临床试验评估了本品与泊马度胺和地塞米松的安全有效性。307名受试者按1:1的比例随机分组,分别在泊马度胺和地塞米松治疗的基础上,增加本品或安慰剂治疗,结果显示本品治疗组总缓解率为60.4%,中位PFS为11.53个月,而安慰剂组分别仅为35.3%和6.47个月,中位OS尚未达到[xxiv]。多发性骨髓瘤是一种较为少见的肿瘤,美国美国国家癌症研究所数据显示,美国2020年将有32270名新发病例和12830名死亡病例[xxv]。在本品获批之前,FDA已经批准了强生的CD38抗体daratumumab,相比isatuximab,daratumumab获准的应用范围更加广泛,而且可以实现皮下注射治疗,是本品最强的竞争对手,EVP预测本品在2024年的销售额为5.16亿美元。10. Isturisa(osilodrostat)3月6日,FDA批准了Recordati公司的osilodrostat片,用于不适宜垂体手术或经过手术未治愈的成年库欣病患者治疗。库欣病是由垂体瘤引起,垂体释放出过多促肾上腺皮质激素引起肾上腺皮质醇合成过量,进而引发一些列的临床症状。库欣病是一种极为罕见的疾病,美国国家罕见病组织数据显示,该病的年发病率仅百万分之十三[xxvi]。Osilodrostat是一种皮质醇合成抑制剂,可抑制11β-羟化酶的活性,阻断肾上腺合成皮质醇。FDA批准本品上市是基于一项有137名患者参与的临床试验(NCT02180217)的结果,经过24周的开放标签治疗,约有一半患者的皮质激素水平下降至正常范围内。在此基础上,71名无需增加治疗剂量(30mg每日2次)且在12周内耐受该药物的患者被纳入为期8周的双盲、随机停药研究,结果显示,继续使用osilodrostat治疗的患者中,86%的患者皮质激素维持在正常水平,而停药改用安慰剂的患者仅为30%[xxvii][xxviii]。市场潜力方面,EVP预测本品在2024年的销售额为4000万美元。
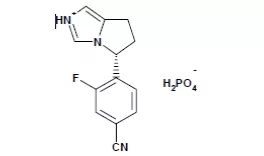
11. Zeposia(ozanimod)3月25日,FDA批准了Celgene的ozanimod,用于治疗成人复发型多发性硬化症(RMS),包括临床孤立综合征、复发缓解性疾病、活动性继发进展性疾病。Ozanimod是一种鞘氨醇1-磷酸(s1p)受体调节剂,与s1p受体1和受体5有非常高的亲和力,但在多发性硬化中的作用机制尚不完全清楚。FDA批准本品上市是基于两项随机、双盲、安慰剂对照的临床研究结果。试验一((NCT02294058)共计有895名患者入组,持续治疗1年,试验二(NCT02047734)共有874名患者入组,持续治疗2年,患者入组后均接受本品和β干扰素-1a治疗,主要终点均为治疗期内的年化复发率。试验一的结果显示,本品治疗组的年化复发率为0.181,78%的患者未出现复发,β干扰素治疗组年化复发率为0.350,66%的患者未出现复发,达治疗终点[xxix];试验二结果显示本品治疗组的年化复发率为0.172,76%的患者未出现复发,β干扰素治疗组年化复发率为0.276,64%的患者未出现复发,也达到了治疗终点[xxx]。除了MS,ozanimod正在开发的适应症包括克罗恩病和炎性肠病,EVP预测本品在2024年的销售额为9.66亿美元。
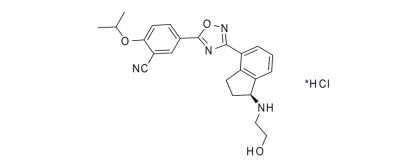
12. Koselugo(selumetinib)4月10日,FDA批准了阿斯利康的selumetinib,用于两岁以上、无法手术的1型神经纤维瘤(NF1)患者治疗。NF1是一种非常罕见的疾病,估计每3000个婴儿中将会出现一个患者,而selumetinib首个批准用于该疾病的药物。Selumetinib是一个丝裂原活化蛋白激酶激酶1/2(MEK1/2)选择性抑制剂,FDA批准本品是基于一项单臂、多中心的2期临床研究结果,该试验共入组了50名患者,平均年龄为10.2岁。经过治疗,33名患者出现完全或部分缓解,总缓解率为66%,27名患者持续缓解时间超过12个月,占总缓解患者的82%[xxxi]。除了NF1,阿斯利康还在开发胶质瘤、甲状腺癌等适应症,EVP预测本品在2026年的销售额可达0.84亿美元。
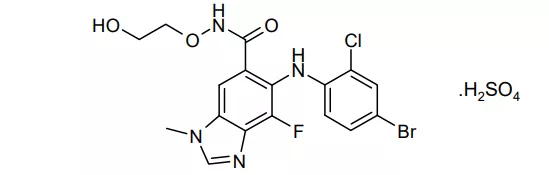
13. Tukysa(Tucatinib)4月17日,FDA批准了Seattle Genetics公司的Tucatinib,适应症是与曲妥珠单抗和卡培他滨联用,治疗晚期无法手术或转移性的HER2(人表皮生长因子受体-2)阳性乳腺癌成人患者,包括既往接受过1种以上HER2治疗方案的脑转移患者。本品的获批是基于一项有612名患者参与的临床研究结果,受试者被2:1随机分组,分别接受本品+曲妥珠单抗+卡培他滨或安慰剂+曲妥珠单抗+卡培他滨治疗,结果显示本品治疗组的中位PFS和中位OS分别为7.8个月和21.9个月,而安慰剂组仅分别为5.6个月和17.4个月,均达到统计学显著差异[xxxii]。HER2阳性患者约占乳腺癌总病例数的五分之一,除了乳腺癌,本品还在开发直肠癌等适应症,EVP预测本品在2026年的销售额为9.50亿美元。 14. Pemazyre(pemigatinib)同在4月17日, FDA批准还加速批准了Incyte公司的成纤维生长因子(FGF)受体2抑制剂pemigatinib,用于携带FGFR2基因融合/重排、既往接受过治疗且不可手术切除或局部晚期的胆管癌成人患者治疗。FDA加速批准本品上市是基于一项名为FIGHT-202 (NCT02924376)的临床试验的结果。这是一项多中心、有107名FGFR2基因融合/重排患者参与的单臂2期临床试验,患者接受本品治疗后,38名患者出现缓解(36%),其中35名患者部分缓解,3名患者完全缓解,中位持续缓解时间是9.1个月,24名患者持续缓解时间超过6个月,7名患者持续缓解时间超过12个月[xxxiii]。胆管癌是一种非常罕见的癌症,美国每年新确诊患者约为8000人[xxxiv],其中且仅有15%的患者属于FGFR2基因突变[xxxv],EVP预测该产品在2026年的销售额为3.35亿美元。
14. Pemazyre(pemigatinib)同在4月17日, FDA批准还加速批准了Incyte公司的成纤维生长因子(FGF)受体2抑制剂pemigatinib,用于携带FGFR2基因融合/重排、既往接受过治疗且不可手术切除或局部晚期的胆管癌成人患者治疗。FDA加速批准本品上市是基于一项名为FIGHT-202 (NCT02924376)的临床试验的结果。这是一项多中心、有107名FGFR2基因融合/重排患者参与的单臂2期临床试验,患者接受本品治疗后,38名患者出现缓解(36%),其中35名患者部分缓解,3名患者完全缓解,中位持续缓解时间是9.1个月,24名患者持续缓解时间超过6个月,7名患者持续缓解时间超过12个月[xxxiii]。胆管癌是一种非常罕见的癌症,美国每年新确诊患者约为8000人[xxxiv],其中且仅有15%的患者属于FGFR2基因突变[xxxv],EVP预测该产品在2026年的销售额为3.35亿美元。
15. Trodelvy(sacituzumab govitecan-hziy)4月22日,FDA加速批准了Immunomedics公司的抗体药物偶联物(ADC)sacituzumab govitecan,用于治疗既往已接受至少2种疗法的转移性三阴性乳腺癌成人患者。转移性三阴乳腺癌约占侵袭性乳腺癌的15%,目前治疗手段仍以化疗为主,但一般化疗效果较差,应答率一般仅为10%-15%,PFS则只有2-3个月。Sacituzumab是一种靶向于TROP-2(人滋养层细胞表面抗原2)的人源化IgG1抗体,而govitecan是拓扑异构酶抑制剂伊立替康的代谢活性产物。本品的安全有效性在一项名为IMMU-132-01 (NCT01631552)的多中心、单臂临床试验中得到初步评估,结果显示,108名接受本品治疗的患者中有36名患者出现缓解(ORR:33.3%),3名患者完全缓解(2.8%),33名患者部分缓解(30.6%),中位持续缓解时间为7.7个月,中位PFS和中位OS分别为5.5个月和13.0个月[xxxvi]。除了三阴乳腺癌,本品开发的适应症还包括直肠癌、前列腺癌、胃癌等多种实体瘤,是2020年最值得关注的“重磅级炸弹”之一,EVP预测该产品在2026年的销售额可达21.51亿美元。16. Ongentys(opicapone) 4月24日,FDA批准了Neurocrine公司的新一代儿茶酚-氧位-甲基转移酶(COMT)抑制剂opicapone,与甲基多巴和左旋多巴联用,以改善帕金森病(PD)患者“关”时间。开关现象是PD患者长期应用左旋多巴出现的疗效波动,而“关”主要表现为突然出现肢体僵直,运动不能,此前获批用于改善开关现象治疗的COMT抑制剂有托卡朋和恩他卡朋,但托卡朋因为肝毒性已经很少用。FDA批准opicapone上市是基于两项随机、双盲的临床研究结果。入组两项临床试验的患者均接受左旋多巴和卡比多巴的背景治疗,试验一(NCT01568073)有600名患者入组,分别接受本品5mg、25mg、50mg或安慰剂添加治疗,结果显示,接受本品50mg治疗的115名患者,平均关闭时间相比基线下降1.95小时,而接受安慰组的120名患者仅平均下降0.93小时,达治疗终点,5mg和25mg组未达治疗终点[xxxvii]。试验二(NCT01227655)的设计与试验一类似,结果显示,接受本品50mg治疗的147名患者,平均关闭时间相比基线下降1.98小时,而接受安慰组的135名患者仅平均下降1.07小时,也达治疗终点,25mg也未达治疗终点[xxxviii]。市场方面,本品早在2016年就获得EMA批准上市,但市场表现平平,EVP预测该产品在2026年的销售额为3.44亿美元。
17. Tabrecta (capmatinib)5月6日,FDA加速批准了诺华公司的capmatinib,用于治疗MET(上皮-间质细胞转化)外显子14跳跃突变的转移性非小细胞肺癌(NSCLC)成人患者。FDA做出这项决定的基础是一项多中心、开放标签的临床研究(NCT02414139)的早期结果,该试验共入组了364名患者,其中97名患者携带了MET外显子14跳跃突变,28人为初治,其余69人为经治。经过本品的治疗,初治患者亚组的总缓解率为68%(4%完全缓解,64%部分缓解),中位DOR为12.6个月,47%缓解患者的持续缓解时间超过12个月,而经治患者亚组的总缓解率为41%(无完全缓解),中位DOR为9.7个月,32%缓解患者的持续缓解时间超过12个月。携带MET外显子14跳跃突变的NSCLC占肺癌患者总量的3%-4%[xxxix],本品是首个获FDA批准的针对性疗法。除了NSCLS,诺华还在开发乳腺癌、淋巴瘤和肾细胞癌等适应症,EVP预测该产品在2026年的销售额可达3.55亿美元。
18. Retevmo (selpercatinib)5月8日,FDA加速批准了Loxo oncology公司的selpercatinib,用于治疗RET(转染重排基因)融合阳性的转移性非小细胞肺癌和甲状腺癌,以及RET基因突变的髓样甲状腺癌成人患者。Selpercatinib是首个获得FDA批准用于RET基因改变的药物,FDA加速批准本品上市是基于一项名为LIBRETTO-001(NCT03157128)的开放标签、且同时针对多种实体瘤的临床研究结果。RET融合的NSCLC方面,108名既往接受过顺铂化疗后进展的患者,经本品治疗后的总缓解率为64%(CR:1.9%,PR:62%),中位DOR为17.5个月,而39名初治患者的总缓解率为85%(CR:0%,PR:85%),中位DOR时间尚未达到[xl]。RET基因突变的髓样甲状腺癌方面,55名既往经历过卡博替尼或凡德他尼治疗后进展的患者,经本品治疗后的总缓解率为69%(CR:9%,PR:60%),中位DOR时间尚未达到,而88名初治患者的总缓解率为73%(CR:11%,PR:61%),中位DOR时间也尚未达到[xli]。RET融合的甲状腺癌方面,既往经受过放射性碘、索拉菲尼或仑伐替尼等药物全身性治疗后进展的19名患者,经本品治疗后的总缓解率为79%(CR:5.3%,PR:74%),中位DOR为18.4个月,而9名初治患者,总缓解率达100%(CR:12.5%,PR:88%),中位DOR时间尚未达到[xlii]。本品的潜在患者群为1%-2%的NSCLC、10%-20%的甲状腺乳头状癌(占甲状腺癌的80%)[xliii]和70%的髓样甲状腺癌(占甲状腺癌的4%)[xliv][xlv],EVP预测本品在2026年的销售额可达11.72亿美元。
19. Qinlock (ripretinib)5月15日,FDA批准了Deciphera制药的ripretinib,用于转移性胃肠间质瘤四线治疗。Ripretinib是一种广谱KIT原癌基因受体酪氨酸激酶抑制剂和PDGFR(血小板衍生生长因子受体)α激酶抑制剂,FDA批准该产品上市是基于一项多中心、随机双盲的临床试验(NCT03353753)的结果。129名经伊马替尼、舒尼替尼或瑞戈非尼等靶向药物治疗后进展的胃肠间质瘤患者,按2:1随机分组,分别接受本品或安慰剂治疗,结果显示本品治疗的85名患者中,9%出现缓解,中位PFS和中位OS分别为6.3个月和15.1个月,而安慰剂组的44名患者中,无人获得缓解,中位PFS和中位OS分别仅为1.0个月和6.6个月[xlvi]。基因检测数据显示,超过85%的胃肠间质瘤和系统性肥大细胞增多症病例检测到KIT或PDGFRA激酶,另外急性髓细胞白血病(AML)、黑色素瘤,生殖细胞肿瘤,肺癌和成胶质细胞瘤也存在一定比例的KIT或PDGFRA激酶[xlvii],因此本品的应用前景比较广阔,EVP预测本品在2026年的销售额可达13.11亿美元。  20. Cerianna (fluoroestradiol F18)5月20日,FDA批准了Zionexa公司的含氟正电子发射型计算机断层显像(PET)剂,用于雌激素受体(ER)阳性病变检测,作为复发或转移性乳腺癌患者活检的辅助手段。本品是一种F-18标记的F取代雌二醇,与雌激素受体有很强的亲和力,而F-18是正电子放射性核素,可核变产生的正电子与周围的负电子发生堙灭现象,产生湮没辐射而成相。FDA批准本品上市是基于一项有93名患者参与的临床试验(NCT01986569)的结果,在完成有效性分析的88名患者中,47名为雌激素受体阳性,38名为雌激素受体阴性。试验通过评估免疫组织化学测定结果与注射 cerianna后PET-CT(正电子发射计算机断层显像)成像结果之间的一致性来判断诊断结果的准确性,结果显示,阳性患者检测结果一致率为76.6%,而阴性患者为100%[xlviii]。
20. Cerianna (fluoroestradiol F18)5月20日,FDA批准了Zionexa公司的含氟正电子发射型计算机断层显像(PET)剂,用于雌激素受体(ER)阳性病变检测,作为复发或转移性乳腺癌患者活检的辅助手段。本品是一种F-18标记的F取代雌二醇,与雌激素受体有很强的亲和力,而F-18是正电子放射性核素,可核变产生的正电子与周围的负电子发生堙灭现象,产生湮没辐射而成相。FDA批准本品上市是基于一项有93名患者参与的临床试验(NCT01986569)的结果,在完成有效性分析的88名患者中,47名为雌激素受体阳性,38名为雌激素受体阴性。试验通过评估免疫组织化学测定结果与注射 cerianna后PET-CT(正电子发射计算机断层显像)成像结果之间的一致性来判断诊断结果的准确性,结果显示,阳性患者检测结果一致率为76.6%,而阴性患者为100%[xlviii]。 21. Artesunate(青蒿琥酯)5月26日,FDA批准了Amivas公司的青蒿琥酯用于成人或儿童严重疟疾患者初始治疗。青蒿琥酯已经在我国使用多年,此次在美国获批是基于两项10年前的研究结果,其中试验一是一项2005年在亚洲开展的开放标签的临床试验,而试验二是2010年在非洲开展的开放标签的临床试验,试验一共有1461名患者入组,而试验二则有5425名患者入组,两项试验均以奎宁作为对照,以院内死亡率作为主要终点。在试验一中,青蒿琥酯(N=730)治疗组的院内死亡率为15%,而接受奎宁(N=731)治疗组为22%[xlix],试验二的结果与试验一类似,其中青蒿琥酯治疗组(n=2712)的院内死亡率为8.5%,而奎宁(n=2713)组为10.9%[l]。疟疾是对人类影响最大的传染病之一,全世界有3至5亿人感染过疟原虫,每年有150至270万人死于该病,其中90%以上病患在非洲[li]。美国CDC(疾病预防和控制中心)数据显示,每年仅有约2000个疟疾确诊病例,其中仅300例为重症患者[lii]。
21. Artesunate(青蒿琥酯)5月26日,FDA批准了Amivas公司的青蒿琥酯用于成人或儿童严重疟疾患者初始治疗。青蒿琥酯已经在我国使用多年,此次在美国获批是基于两项10年前的研究结果,其中试验一是一项2005年在亚洲开展的开放标签的临床试验,而试验二是2010年在非洲开展的开放标签的临床试验,试验一共有1461名患者入组,而试验二则有5425名患者入组,两项试验均以奎宁作为对照,以院内死亡率作为主要终点。在试验一中,青蒿琥酯(N=730)治疗组的院内死亡率为15%,而接受奎宁(N=731)治疗组为22%[xlix],试验二的结果与试验一类似,其中青蒿琥酯治疗组(n=2712)的院内死亡率为8.5%,而奎宁(n=2713)组为10.9%[l]。疟疾是对人类影响最大的传染病之一,全世界有3至5亿人感染过疟原虫,每年有150至270万人死于该病,其中90%以上病患在非洲[li]。美国CDC(疾病预防和控制中心)数据显示,每年仅有约2000个疟疾确诊病例,其中仅300例为重症患者[lii]。
22. Tauvid(flortaucipir F 18)5月28日,FDA批准了Avid radiopharm的Tauvid,用于τ蛋白神经纤维纠结的密度和分布成像,以为阿尔茨海默病(AD)诊断提供评估依据。Flortaucipir F 18可与聚集的τ蛋白结合,靶向分布在病变区域。Tauvid的安全有效性在两项临床试验中进行了评估,其中试验一(NCT02516046)入组了156名同意tauvid成像并死后接受大脑捐赠的绝症患者,其中64名患者完成了成像结果与大脑病理切片的一致性评估,结果显示,5个独立读片者的读片灵敏度为92%,特异性在52%-92%之间。试验二(NCT03901092)在试验一中156名患者的基础上增加了159名因认知障碍而确诊为AD的患者,使用属性型MSA分析(Fleiss’ kappa)统计五名读片者的读取结果一致性,结果显示241名不同患者的读取结果一致性为0.87,其中绝症患者的读取结果一致性为0.82,而确诊患者为0.90[liii]。使用PET技术对病理性τ成像,将有助于AD早期诊断和疾病进展情况监测,在提倡AD疾病早期干预的时代大背景下,τ蛋白和β-淀粉样蛋白示踪剂都具有巨大的发展潜力,是国外诊断巨头们争相布局的领域之一。
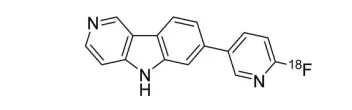
23. Uplizna (inebilizumab-cdon)6月11日,FDA批准了Viela生物的inebilizumab,用于水通道蛋白4 (AQP4)抗体阳性的视神经脊髓炎谱系疾病(NMOSD)治疗。NMOSD是一种非常罕见的疾病,患者的免疫系统会错误地攻击体内的健康细胞和蛋白质,大约50%的患者会发展成为永久性视力障碍和瘫痪。流行病学数据显示,约有1.6万-1.7万美国人罹患此病[liv]。在一项有230名NMOSD患者参与的临床试验(NCT02200770)中,213名患者被确定为AQP4抗体阳性。AQP4抗体阳性患者被2:1随机分两组,分别接受Uplizna或安慰剂治疗。在为期197天的研究中, Uplizna治疗组的NMOSD复发率为11.2%,而安慰剂治疗组的复发率为42.3%,无证据表明AQP4抗体阴性患者在治疗中受益[lv]。除了NMOSD,本品还在开发肾移植、重症肌无力等多种自免疫疾病,EVP预测该产品在2026年的销售额可达5.18亿美元。24. Zepzelca(lurbinectedin)6月22日,FDA加速批准了Jazz制药的lurbinectedin,用于铂类化疗中进展或化疗后进展的晚期小细胞肺癌(SCLC)治疗。小细胞肺癌约占肺癌总数的13%-15%,目前治疗仍以化疗为主。Lurbinectedin是一种新型烷化剂,FDA提前批准该产品上市是基于一项名为Study B-005(NCT02454972)、开放标签的临床研究结果。105名铂类化疗中进展或化疗后进展的晚期SCLC患者经过本品治疗后,总缓解率为35%(无完全缓解),其中45名铂类耐药患者(最后一剂治疗后90天内复发)亚组ORR为22%,而60名铂敏感患者亚组的ORR为45%;中位DOR为5.3个月,铂耐药和铂敏感亚组分别为4.7个月和6.2个月[lvi]。在二线治疗的基础上,lurbinectedin还有潜力用于一线治疗,在上述临床试验中,对铂类化疗后180天内无复发的20名患者单独分析显示,ORR为60%,中位PFS和中位OS分别为4.6个月和16.2个月,NCCN(美国国立综合癌症网络)建议Jazz对这一类患者开展一线治疗的研究[lvii]。市场潜力方面,EVP预测本品在2026年的销售额可达3.97亿美元。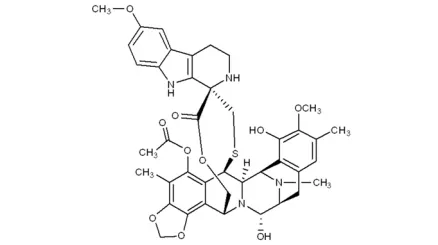
25. Dojolvi (三庚酸甘油酯)6月30日,FDA批准了Ultragenyx公司的三庚酸甘油酯作为能量和脂肪酸的来源,用于长链脂肪酸氧化紊乱(LC-FAOD)患者治疗。LC-FAOD是一种非常罕见而危及生命的常染色体隐性遗传病,美国每年约有100名新生儿被确诊为该病,共计有2000-3500名存活患者[lviii]。该病患者因无法将长链脂肪酸转化为能量而导致线粒体能量生成不足,临床表现为心肌病、运动不耐受、频繁肌痛,反复横纹肌溶解和低血糖等。本品是一种中链脂肪酸甘油酯,其安全有效性在一项为期4个月的随机、双盲的临床试验(NCT01379625)中得到了评估,32名LC-FAOD受试患者,分别接受本品(7碳)和三辛酸甘油酯(8碳)治疗4个月,结果显示,两组间关键指标无显著差异,每个组均有7次横纹肌溶解事件[lix],除了LC-FAOD,本品还在开展多项代谢失调的适应症研究,EVP预测其在2026年的销售额为2.68亿美元。
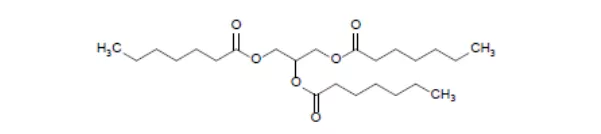
26. Rukobia (fostemsavir)7月2日,FDA批准了全新机制的抗逆转录药物fostemsavir,适应症是与其他抗逆转录药物联用,治疗多重耐药或因耐药性、不耐受或安全性问题而既往接受过大量现有方案治疗失败的成人HIV-1(1型人类免疫缺陷病毒)感染者。Fostemsavir是一种由葛兰素史克(GSK)开发上市的病毒附着抑制剂,其安全有效性已在一项随机、双盲的三期临床试验(NCT02362503)中得到评估,371名HIV RNA(核糖核酸)载量≥400拷贝/ml的多重耐药患者加入了临床试验,其中272名患者因存在1-2种完全敏感的市售抗逆转录制剂而被分配到组1,而其余99名患者因无完全敏感的市售抗逆转录制剂而被分配到组2。组1的患者在最佳背景治疗方案的基础上,随机分为2个亚组,分别接受fostemsavir(n=201)或安慰剂治疗(n=69)8天,然后所有患者均使用fostemsavir,进行开放标签治疗。结果显示,fostemsavir治疗组患者,在第8天的病毒RNA载量相比第1天下降了0.791个log10值,而安慰剂组仅下降了0.166个log10值[lx],达统计学显著差异,开放标签治疗后的第24周和第96周分别有53%和60%的患者,体内病毒RNA载量低于40拷贝/ml。被分配到组2的99名患者直接接受开放标签治疗,第24周和第96周均有37%的患者,体内病毒RNA载量低于40拷贝/ml[lxi]。市场潜力方面,本品因剂量太大,难以开发成每日一片的鸡尾酒,EVP预测本品在2026年的销售额为2.68亿美元。
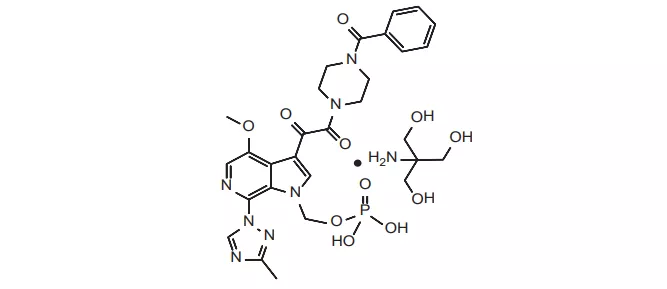
27. Byfavo(瑞马唑仑)7月2日,FDA还批准了Acacia公司的瑞马唑仑,用于时长不超过30分钟的成人手术的镇静诱导或维持。瑞马唑仑是一种超短效苯二氮卓类药物,FDA批准本品上市主要是基于三项临床试验的结果。试验一(NCT02290873)入组了461名ASA-PS(美国麻醉师协会患者体质评分)为1-3级的肠镜检查受试者,其中298名受试者给予了瑞马唑仑,60名患者给予了安慰剂,两组患者都在镇静剂给药前给予芬太尼,并且研究人员可以根据具体情况判断是否给予咪达唑仑紧急镇静治疗。以“顺利完成肠镜检查”、“无需紧急镇静治疗药物”和“15min窗口期内所需研究药物不超过5剂”为综合指标,评价药物的镇静成功率,结果显示瑞马唑仑治疗组的镇静成功率为91.3%,而安慰剂组仅为1.7%[lxii]。试验二的((NCT02296892)设计和评估标准与试验一相同,入组的431名ASA-PS为1-3级的支气管镜受试者,结果显示,本品和安慰剂的镇静成功率分别为80.6%和4.8%[lxiii]。试验三((NCT02532647)是一项针对ASA-PS为3-4级肠镜受试者设计的临床试验,该试验的设计几乎与试验一相同,只是给药剂量较大,主要目的是评估产品的安全性。试验结果显示,两组受试者镇静成功率分别为84.4%和0%,两组无严重不良反应,低血压发生率分别为61.3%和75%[lxiv]。本品除了在支气管镜、肠镜、胃镜[lxv],在手术全麻方面,也有一项2期临床试验证明,本品的疗效不劣于丙泊酚[lxvi],EVP预测本品在2026年的销售额可达2.69亿美元。
28. Inqovi (地西他滨 + cedazuridine)
7月7日,FDA批准了大冢制药的固定剂量组合物Inqovi,用于治疗成人骨髓增生异常综合征(MDS)患者。Inqovi由35mg的抗核酸代谢药物地西他滨和100mg的胞苷脱氨酶抑制剂cedazuridine组成,苷脱氨酶抑制剂可抑制胞苷类药物(含地西他滨)在胃肠道降解,以提高生物利用度水平。FDA批准本品上市的基于两项开放标签、双周期、双交叉临床试验的研究结果,试验一(NCT02103478)共计有80名MDS或慢性粒单核细胞白血病(CMML)患者入组,按1:1随机给予Inqovi或20mg的地西他滨注射治疗,经过28天一疗程的治疗后,第二疗程交换用药方案,在第二个疗程之后则全部患者接受本品开放标签治疗,经过多个疗程的治疗后,总缓解率为60%,完全缓解率为21%,中位DOR为13.3个月[lxvii]。试验二(NCT03306264)与试验一的设计相似,133名受试者在多个疗程的治疗后,完全缓解率为21%,中位DOR为7.5个月[lxviii]。MDS是一种较为罕见的疾病,美国的年龄调整发病率为十万分之四,五年生存率仅为31%,EVP预测本品在2026年的销售额为0.77亿美元,然而cedazuridine的价值不限于此,还有望与阿扎胞苷等多种胞苷类药物联用,以实现口服给药。
29. Xeglyze(Abametapir)7月24日,FDA批准了Dr Reddy's公司的abametapir洗剂,用于6个月及以上年龄段人群的头虱感染治疗。Abametapir是一种金属蛋白酶抑制剂,可影响头虱存活或虫卵发育。FDA批准abametapir是基于2项多中心、随机双盲、安慰剂对照的临床研究结果。两项试验共计有704名受试者入组,其中216名为家庭性感染(3人以上感染)中的最年轻个体被指定为索引个体(index subjects),疗效评估标准为受试者接受本品或安慰剂单次治疗后第1、7、14天内受随访的索引个体无活虱的比例,如果索引个体在1-14天内随访中发现有活虱则意味着治疗失败。结果显示,81.5%的abametapir治疗患者在14天内随访中无活虱,而安慰剂组仅为49.1%。合并非索引个体一起统计,704名受试者中,abametapir治疗组有85.9%的患者14天内无活虱,而安慰剂治疗组仅为61.3%,最常见的不良反应是皮肤潮红(4.0%)、皮疹(3.2%)和皮肤灼热感(2.6%)[lxix]。尽管杀虫剂已经广泛使用,但现代社会头虱感染依然非常普遍,尤其是儿童,据美国CDC数据显示,每年有600-1200万名3-11岁的儿童感染头虱[lxx]。
30. Monjuvi (tafasitamab-cxix)
7月31日,FDA加速批准了MorphoSys公司的tafasitamab,适应症是与来那度胺联用,治疗复发或难治性的弥漫性大B细胞淋巴瘤(DLBCL)成人患者。FDA批准本品提前上市是基于一项开放标签的多中心临床研究(NCT02399085)的结果,71名入组患者均既往经历过1-3种全身性治疗方案治疗,经过本品治疗后,39名患者出现缓解,总缓解率为55%,37%的患者CR,18%的患者PR,中位DOR为21.7个月[lxxi]。DLBCL是最常见的非霍奇金淋巴瘤(NHL)亚型,据美国NIH公开的数据显示,DLBCL的年发病率为5.6/10万,五年生存率为63.8%[lxxii],每年约有2万名新确诊患者。Tafasitamab是一种Fc片段优化的CD19单抗,而CD19为多种B细胞恶性肿瘤的生物标记物,因此除了DLBCL,tafasitamab还有望用于慢性髓细胞淋巴瘤(CLL)、NHL和前体B细胞淋巴细胞白血病等适应症,EVP预测该产品在2026年的销售额将达6.87亿美元。31. Blenrep(belantamab mafodotin-blmf)8月5日,FDA加速批准了GSK的ADC药物belantamab mafodotin,用于既往至少接受过4种疗法的复发/难治性多发性骨髓瘤(MM)成人患者治疗。Belantamab是一种以B细胞成熟抗原(BCMA)为导向的单克隆抗体,小分子的部分是一种微管蛋白抑制剂。FDA批准本品是基于一项多中心、开放标签的双臂临床研究(NCT 03525678)的早期结果,目前该试验还在进行中。该试验共计筛选了293名患者,其中193名患者被纳入到意向治疗人群,97名患者被分配到2·5 mg/kg剂量组,99名分配至3·4 mg/kg剂量组,结果显示,30名2·5 mg/kg剂量组患者和34名2·5 mg/kg剂量组患者出现缓解,最常见的3-4级不良反应主要是角膜病变、血小板减少和贫血,两名患者因治疗相关的原因死亡[lxxiii]。虽然全球大约只有1万MM接受四线治疗,但GSK还在开展更早期的用药试验,EVP预测该产品在2026年的销售额可达12.62亿美元。32. Lampit (nifurtimox)8月6日,FDA加速批准了拜耳的nifurtimox片,用于儿童查加斯病治疗。Nifurtimox的安全有效性在一项于拉美地区开展的临床试验(NCT02625974)中得到了评估,330名受试者按2:1的比例被随机分配到60天治疗组和30天治疗组,按体重给予nifurtimox治疗,随访一年,并以“裂解液抗原酶联免疫吸附测定结果(Lysate ELISA)、重组抗原酶联免疫吸附测定结果(Recombinant ELISA)下降20%以上”,或“一年后血清免疫球蛋白转阴”为综合指标评估血清学应答率。结果显示,60天治疗组以Lysate ELISA计算的血清应答率为32%,以Recombinant ELISA计算的血清应答率为35%,30天治疗组对应的血清应答率则分别仅为19%和22%[lxxv]。查加斯病的病原体为克氏锥虫(trypanosoma cruzi),据世界卫生组织估计,全球有800万人感染了该查加斯病病原体,主要集中在拉美地区,据估计,可能有30万美国人已感染病原体[lxxvi]。除了本品,仅有benznidazole获批用于该病,但治疗获益不理想,尚存在巨大的为满足需求。
33. Olinvyk(oliceridine)8月7日,FDA批准了Trevena公司的oliceridine,用于需要静脉注射阿片药物的急性疼痛管理。Oliceridine是新一代阿片药物,对μ受体上的G蛋白具有选择性激动作用,对β-arrestin的影响较少,FDA批准此项疗法的主要依据是两项临床试验的结果。试验一(NCT02815709)共入组了389名在骨外科-骨切除术后出现中度至重度急性疼痛的患者,在麻醉结束后分别接受安慰剂、oliceridine 0.1mg、oliceridine 0.35mg、oliceridine 0.5mg和吗啡治疗,用“48小时疼痛强度评分总差和(SPID-48)相比基线下降30%以上”、“无需接受紧急救治”、“未在治疗结束前退出”和“用药剂量未达上限”为综合指标计算应答率,其中SPID-48定义为48小时内每个时间点的疼痛强度评分差值(相比基线)乘以所持续时间所得结果的总和。统计结果显示,三个oliceridine治疗组的应答率分别为50.0%、62.0%、65.8%,其中0.35mg 和0.5mg剂量组与吗啡达到非劣标准[lxxvii]。试验二(NCT02820324)入组了401名整形外科-腹部整形术后出现中重度疼痛的患者,试验的设计与试验一相似,但疗效评估时间缩减至24小时。结果显示,五个治疗组的应答率分别为45.7%、61.0%、76.3%、70.0%和78.3%,与安慰组相比,0.35mg和0.5mg组均产生统计学显著差异,而且与吗啡治疗组达到非劣标准[lxxviii]。市场潜力方面,EVP预测该产品在2026年的全球销售额将达2.26亿美元。
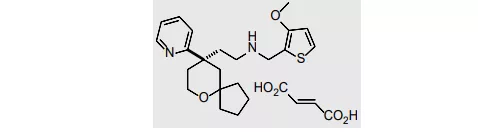
34. Evrysdi(risdiplam)8月7日,FDA一同批准了基因泰克的risdiplam,用于2个月及以上年龄段人群的脊髓性肌萎缩症(SMA)治疗。Risdiplam是一种运动神经元生存基因2(SMN2)剪接修饰剂,可持续增加并维持SMN蛋白的水平,FDA批准该产品上市是基于两项临床研究的结果。试验一(NCT02913482)是一项开放标签的临床试验,21名受试者均为出生后28天-3个月内出现症状的1型SMA患儿,分别接受高剂量(n=17)和低剂量(n=4)的risdiplam治疗,有效性的评估标准是“在没有支撑的情况下能够持续坐立5秒”和“在无需持久通气的条件下存活”。经过12个月的治疗,41%的高剂量组患者能够持续坐立超过5秒,90%入组患者均能在不持久通气的条件下存活。试验二(NCT02908685)是一项针对2型和3型SMA患者开展的临床试验,该试验分成了两个部分,第一个部分是剂量探索性试验,第二个部分是安慰剂对照的随机双盲试验,其疗效评估指标为第12个月的MNM32(Motor Function Measure 32)量表评分与基线的差值。结果显示接受risdiplam治疗的120名患者,平均MNM32量表评分相比基线增加1.36分,而安慰剂组则下降了0.19分,达统计学显著性差异[lxxix]。SMA是儿童期最常见的常染色体隐性遗传病之一,数据显示I型、II型和III型分别占60%、27%和12%,发病率分别为5.5/10万、1.9/10万和1.7/10万[lxxx],EVP预测本品在2026年的销售额将达17.86亿美元。
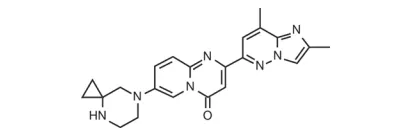
35. Viltepso(viltolarsen)8月12日,FDA加速批准了NS Pharma公司的反义寡核苷酸viltolarsen,用于DMD(杜氏肌营养不良症)基因53号外显子跳跃突变的杜氏肌营养不良症治疗。FDA批准本品上市是基于一项多中心、两阶段的剂量相关性研究(NCT02740972)的结果。16名受试者分别随机接受本品或安慰剂治疗4周,然后患者均接受两个不同剂量的viltolarsen进行开放标签治疗20周,以第25周肌营养不良蛋白水平相对于基线的变化情况来评价治疗效果。结果显示,接受本品80mg/kg每周一次治疗的8名患者,通过Western blot方法测定的平均肌营养不良蛋白水平从基线时正常值的0.9%升高至正常值的5.9%,而使用质谱法测定的平均肌营养不良蛋白水平从基线时正常值的0.6%升高至正常值的4.2%,达到预先设定的试验终点。DMD是一种X连锁的遗传病,整体患病率为2.8/10万,DMD患者的肌营养不良蛋白水平通常不足正常值的3%,大部分患者因呼吸衰竭和心脏功能障碍而死亡[lxxxi],[lxxxii]。虽然此前FDA已经批准了eteplirsen、golodirsen和地夫可特等药物,但临床需求依然未得到良好的满足,EVP预测本品在2026年的销售额可达5.85亿美元。
36. Enspryng(satralizumab)8月14日,FDA批准了基因泰克的IL-6R (白介素-6受体)单抗satralizumab,用于AQP4抗体阳性的视神经脊髓炎谱系疾病治疗。Satralizumab的获批是基于两项为期96周的临床研究结果。试验一(NCT02073279)入组了95名患者,其中64名患者AQP4为阳性,41名患者接受了本品治疗,另外23名接受了安慰剂治疗,试验二(NCT02028884)入组了76名患者,其中52名患者AQP4为阳性,26名患者接受了本品治疗,另外26名接受了安慰剂治疗。治疗结果显示,试验一中两组患者的NMOSD复发率分别为22.0%和56.5%,相比安慰剂,satralizumab治疗组复发风险降低了74%[lxxxiii],而试验二的结果与试验一类似,两组患者的NMOSD复发率分别为11.5%和42.3%,相比安慰剂,satralizumab治疗组复发风险降低了78%,均达到统计学显著性差异。两项临床试验均无证据证明AQP4阴性的患者在治疗过程中有获益[lxxxiv]。由于satralizumab是2020年第二个获批NMOSD的产品,市场预期相对第一款将有所下调,EVP预测该产品在2026年的销售额将达4.19亿美元。
37. Winlevi(clascoterone)8月26日,FDA批准了Cassiopea公司的乳膏,用于12周岁以上年龄段人群的寻常痤疮治疗。Clascoterone是一种雄性激素受体抑制剂,FDA批准该产品上市是基于两项多中心、随机双盲的临床研究结果。试验一(NCT02608450)和试验二(NCT02608476)共计招募了1440名患者,按1:1随机分组并接受Cassiopea乳膏或安慰剂治疗12周,以治疗成功率为指标评估产品的疗效,而治疗成功率定义为IGA(研究者整体评价法)评分为0分或1分或IGA评分相比基线值下降不低于2分的患者比例。结果显示,clascoterone在两项试验中的治疗成功率分别为18.4%(试验一)和20.3%(试验二),而对应的安慰剂组的治疗成功率仅分别为9.0%和6.5%,均达到主要治疗终点。痤疮是非常普遍的疾病,全球共计有6.4亿人受痤疮困扰,患者主要是青少年[lxxxv]。市场潜力方面,EVP预测该产品在2026年的销售额将达4.2亿美元。
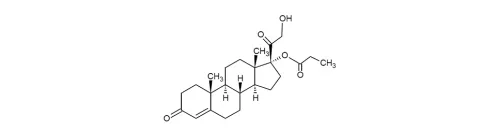
38. Sogroya(somapacitan-beco)
8月28日,FDA批准了诺和诺德每周一次的长效生长激素类似物,以替代内源性生长激素,用作成人生长激素缺乏症(GHD)治疗。Somapacitan是一种脂肪酸修饰的重组生长激素,可通过脂肪链与体内白蛋白结合,以降低清除率。FDA批准somapacitan上市,主要是基于一项为期35周、随机、双盲、安慰剂对照的临床研究结果。入组的GHD初治患者按2:1:2被随机分成三组,分别接受somapacitan(n=120)每周一次(n=60)、安慰剂和普通生长激素每日一次(n=119)治疗34周。结果显示,somapacitan、安慰剂和普通生长激素三个治疗组的躯干脂肪百分率分别相比基线下降1.16%、-0.63%和2.47%。somapacitan、安慰剂和普通生长激素三个治疗组平均胰岛素样生长因子-1标准偏差评分(IGF-I SDS)相比基线分别改善2.37分、0.02分和2.30分[lxxxvi]。虽然激素缺乏症是一种比较罕见的疾病[lxxxvii],但需要长期维持治疗,而顺应性较好的长效生长激素是主要发展趋势,EVP预测somapacitan在2026年的销售额可达3.26亿美元。
39. Detectnet(Cu 64 Dotatate)9月3日,FDA批准了Radio Medix 公司PET显像剂Detectnet,用于生长抑素受体阳性神经内分泌肿瘤(NETs)的定位。Cu 64是一种正电子放射性核素,通过1,4,7,10-四氮杂环十二烷-1,4,7,10-四羧酸(简称DOTA)耦合并链接到生长抑素类似物上,进而选择性地与生长抑素受体2结合,实现病变部位显像。FDA批准Detectnet上市是基于两项临床试验的结果。试验一评估了63名受试者,其中包括42名根据组织学、常规影像学和临床评估判定为NET确诊或疑似患者和21名健康志愿者。三位独立读片人的读片结果与复合标准诊断结果相比较,阳性一致率均为91%,而阴性一致率分别为97%、80%和90%[lxxxviii]。试验二入组了113名受试者,其结果与试验一类似[lxxxix]。
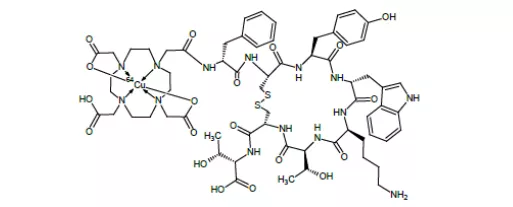
40. Gavreto(pralsetinib)9月4日,FDA加速批准了Blueprint Medicines公司的pralsetinib用于RET融合阳性的成人转移性NSCLC治疗。Pralsetinib是一种RET激酶抑制剂,FDA提前批准该产品上市是基于一项多中心、开放标签的临床研究(NCT03037385)的早期结果。87名既往接受过铂类化疗的转移性RET融合阳性的NSCLC患者,接受本品治疗后观察到的总缓解率为57%,其中CR为5.7%,PR为52%,80%获缓解患者的DOR超过6个月;27名初治患者接受本品治疗后观察到的总缓解率为70%,其中CR为11%,PR为59%,58%的获缓解患者的DOR超过6个月,中位持DOR为9.0个月[xc]。Pralsetinib是2020年度第二个获批用于RET阳性NSCLC的药物[xci],尚在开发阶段的适应症主要是甲状腺癌,EVP预测该产品在2026年的销售额可达1.09亿美元。
41. Inmazeb (atoltivimab, maftivimab, odesivimab-ebgn)10月14日,FDA批准了再生元的抗Zaire埃博拉病毒新药Inmazeb,该产品由三种可抑Zaire埃博拉病毒的抗体组成,FDA批准其上市是基于一项由美国国家过敏和传染病研究所(NIAID)资助的多中心临床研究(NCT03719586)的结果,该试验在刚果共和国招募到681名受试者,患者入组后被均分四组,分别接受本品、MAb114(Ebanga)、瑞德西韦和ZMapp治疗,治疗终点为28天的死亡率差异。结果显示,接受本品治疗的154名患者,在28天内有52名患者死亡,死亡率为34%,其中高病毒载量亚组(基线时,下同)死亡率为64%,低病毒载量亚组死亡率为11%,相比之下,控制组(ZMapp治疗组)的153名患者有78名死亡,死亡率为51%,其中高病毒载量亚组死亡率为88%,低病毒载量亚组为22%[xcii]。虽然Inmazeb的疗效并不完全令人满意,但实现了埃博拉治疗史上的从“无”到“有”。
42. Veklury(瑞德西韦)10月22日,FDA批准了吉利德的瑞德西韦,用于12岁及以上年龄段需要住院的2019年新冠病毒(COVID-19)感染者治疗。FDA批准瑞德西韦上市是基于三项临床研究的结果,试验一(NCT04280705)是由NIAID开展的临床试验,1062名轻中重度患者在标准护理的条件下接受瑞德西韦或安慰剂治疗,试验终点为29天内患者恢复(无需通氧及持续的医疗服务或患者出院)时间。结果显示,瑞德西韦治疗的541名患者,中位恢复时间为10天,而安慰剂组的521名患者,中位恢复时间为15天。两组轻中度患者中位恢复时间均为5天,而重度患者分别为11天和18天[xciii]。试验二(NCT04292899)是一项随机、开放标签、多中心的临床试验,该试验比较了重症患者分别接受瑞德西韦5天疗程(n=191)和10天疗程(n=200)的疗效差异。结果显示,在调整两组间基线差异之后,两组患者第14天的临床状态改善程度相似,死亡率和恢复率无显著差别[xciv]。试验三(NCT04292730)与试验二设计类似,该试验比较了患者分别接受瑞德西韦5天疗程(n=197)、10天疗程(n=193)和标准护理(n=200)后第11天的临床状态差异。结果显示,与仅接受标准护理的患者相比,瑞德西韦5天疗程组临床状态显著性改善,10天疗程组的改善程度未达到统计学显著性差异[xcv]。尽管瑞德西韦已经日本和欧盟批准,但该产品的疗效依然饱受争议[xcvi],WHO通过大数据分析认为该产品对住院患者帮助甚微或没有帮助[xcvii]。不过质疑归质疑,吉利德三季度财报显示,该产品在2020年前三季度的销售额就已达8.73亿美元[xcviii],全年销售额极可能超过10亿美元。
43. Zokinvy(Lonafarnib)11月20日,FDA批准了Eiger Biopharm公司的Lonafarnib,用于降低Hutchinson-Gilford早衰综合症(HGPS)死亡风险,或杂合子LMNA基因突变与纯合子/复合杂合子ZMPSTE24基因突变所致的加工缺陷型类早衰核纤层蛋白病治疗。HGPS是非常罕见的疾病,患病率约为二千万分之一,患病情况无种族、性别、地域差异。该病由LMNA突变产生法尼基化异常的早衰蛋白引起,最终主要因心衰死亡,平均死亡年龄为14.6岁[xcix]。Zokinvy是一种口服法尼基转移酶抑制剂,可预防缺陷型早老蛋白或早老蛋白样蛋白聚集。FDA批准该产品是基于两项开放标签的2期临床研究的结果。试验一(NCT00425607)共计入组了28名患者,包括26名为典型HGPS患者,1名非典型HGPS患者和1名LMNA突变性早老蛋白沉积患者,经过24-30个月的lonafarnib治疗后,27名患者完成了统计分析。在完成试验一之后,26名患者又入组到试验二(NCT00916747),继续接受本品治疗,除此以外,该试验还新入组了35名初治患者(34名典型HGPS患者和1名非典型HGPS患者)。合并两项临床试验的62名患者的死亡率情况进行回顾性分析结果显示,接受Zokinvy治疗的前3年里,平均寿命比未接受治疗的患者延长了0.24年,死亡风险比为0.30。长达11年的随访结果显示,接受Zokinvy治疗的患者,平均寿命比未接受治疗的患者延长了2.5年,死亡风险比为0.40[c]。市场潜力方面,EVP预测该产品在2026年的销售额可达0.99亿美元。
44. Oxlumo (lumasiran)11月23日,FDA批准了Alnylam公司的RNAi药物lumasiran,用于1型原发性高草酸尿症(PH1)治疗。原发性高草酸尿症(PHs)是由编码丙氨酸-乙醛酸氨基转移酶的基因突变所引起的罕见遗传病,在西方国家的患病率为十五万分之一。基因缺陷所致的丙氨酸-乙醛酸氨基转移酶缺乏,可引起草酸生成异常升高,而Lumasiran是一种干扰RNA,可沉默编码HAO1(乙醇酸氧化酶)的mRNA,HAO1缺失可导致从乙醇酸到乙醛酸的转化过程受阻,进而影响草酸的合成[ci]。FDA批准lumasiran上市,是基于两项临床试验的结果。试验一是一项针对6岁以上年龄段人群开展的,随机、双盲、安慰剂对照的临床试验,主要终点为24小时内尿液中的总草酸含量变化。结果显示,26名接受lumasiran治疗的患者,平均尿液中总草酸含量相比基线下降了65%,而13名接受安慰剂治疗的患者仅平均下降了13%。经过6个月的治疗,62%的lumasiran治疗患者24小时总尿酸含量达正常水平,而安慰剂组无一患者达到正常水平。试验二是一项开放标签的临床试验,主要针对6岁以下儿童设计,16名患者经过lumasiran为期6个月的治疗后,平均尿液总草酸含量相比基线下降了71%[cii]。Lumasiran是首个获批用于该疾病治疗的药物,EVP预测该产品在2026年的销售额为2.53亿美元。
45. Imcivree(Setmelanotide)11月25日,FDA批准了Rhythm Pharma的多肽药物setmelanotide,用于前阿黑皮素原(POMC)、前蛋白转化酶枯草溶菌素1(PCSK1)或瘦素受体(LEPR)基因缺陷所致肥胖的6岁以上儿童或成人患者的慢性体重管理。Setmelanotide是一种选择性黑皮质素(MC)4受体激动剂,大脑中的MC4受体参与饥饿,饱腹感和能量消耗调节,而POMC、PCSK1和LEPR缺陷所致的肥胖,被认为与MC4受体激活不足有关。FDA批准该产品上市是基于两项临床试验(NCT02896192和NCT03287960)的研究结果,两项试验均是为期一年的开放标签试验,入组患者体重指数(BMI)均不低于30,试验一统计分析了10名POMC 或 PCSK1缺陷患者,而试验二统计了11名LEPR缺陷患者,结果显示,试验一中8名患者体重下降(相比基线,下同)超过10%(80.0%),而试验二中有5名患者体重下降超过10%(45.5%),两项试验中,患者的平均体重下降幅度分别为23.1kg和9.7kg,平均每日饥饿评分分别下降5.5分和4.4分[ciii]。文献显示POMC、PCSK1和LEPR基因缺陷的早发型严重肥胖患者不足5%[civ],但市场潜力依然巨大,EVP预测该产品在2026年的销售额可达9.55亿美元。
46. Danyelza (naxitamab-gqgk)同在11月25日,FDA还加速批准了Y-Mabs therap 公司的naxitamab,适应症是与粒细胞-巨噬细胞集落刺激因子(GM-CSF)联用,治疗骨骼和骨髓复发或难治性高危神经母细胞瘤患者(一岁及以上)。Naxitamab是一抗神经节苷脂GD2抗体,FDA批准该产品上市是基于两项开放标签的临床试验的结果,两项试验的受试者均为既往经历过至少一种全身性疗法治疗失败或复发的患者,在GM-GSF的联合治疗下,naxitamab均按每疗程9mg/kg的剂量给药治疗。在Study 201 (NCT03363373)中,22名患者接受治疗后,总缓解率为45%,其中36%的患者达CR,9%的患者PR,中位DOR为6.2个月,而在Study 12-230 (NCT01757626)中,38名患者接受治疗后,总缓解率为34%,其中26%的患者达CR,8%的患者PR[cv]。神经母细胞瘤是儿童时期非常常见的恶性肿瘤,15岁以下儿童中的发病率为10.2/100万[cvi],每年约有1500名欧洲儿童确诊为神经母细胞瘤,占儿童肿瘤的8%-10%[cvii]。除了神经母细胞瘤,本品在开发的适应症主要是骨肉瘤,EVP预测该产品在2026年的销售额为2.47亿美元。
47. Gallium Ga 68 PSMA-11(PSMA-11 Ga 68)12月1日,FDA批准了加州大学洛杉矶分校开发的PET成像剂,用于前列腺特异膜抗原(PSMA)阳性的前列腺癌患者的病灶转移或复发情况分析。Ga 68是一种正电子放射性核素,通过络合反应链接到可与PSMA特异性结合的载体上,FDA批准其上市是基于两项开放标签的前瞻性临床试验的研究结果。在第一项试验中,325名组织活检确诊为前列腺癌,且被认为高转移风险,可能需要手术切除前列腺和盆腔淋巴结的患者加入了临床试验。所有患者都接受了本品成像,并使用CT或核磁(MRI)扫描。其中的138名患者接受了前列腺切除术和盆腔淋巴癌清除手术,并获得了组织病理性数据(可评估患者)。抽取6位独立读片人中的3个成员的分析结果与病理学评估结果进行比对,结果显示造影诊断的阳性预测值为61%,阴性预测值为84%,诊断灵敏度为47%,特异性为90%。第二项临床试验招募了635名经过特定治疗后有复发生化证据的前列腺癌患者(手术或放疗后前列腺特异抗原升高)。基于使用本品成像后,CT/MRI扫描的结果, 469名(74%)患者体内至少存在一个病灶区域,其中210名患者在12个月内收集到标准诊断证据(可分析患者),结果显示192名(91%)患者至少存在一个以上的真阳性病灶区域,9位独立读片人的读片阳性率在82%到97%之间[cviii]。
48. Orladeyo(berotralstat)12月3日,FDA批准了BioCryst公司的berotralstat,用于12岁以上儿童或成人遗传性血管性水肿发作预防。Berotralstat是一种口服血浆激肽释放酶抑制剂,FDA批准本品是基于一项随机、双盲、安慰剂对照的临床研究(NCT3485911)结果,120名12岁以上的儿童或成人患者加入了临床试验,他们在长达8周的导入期内至少经历了两次发作,平均为2.9次/月。进入试验后,分别接受本品110mg、150mg或安慰剂治疗24周。试验结果显示,接受本品110mg预防的41名患者在28天内的平均发作频率为1.65次,接受150mg预防的40名患者的平均发作频率为1.31次,而安慰剂组的40名患者为2.35次,相比安慰剂组,两个剂量组的发作频率分别下降了30.0%和44.2%,达统计学显著性差异[cx]。HAE是一种罕见而致命的疾病,每6.7万人中就有一人罹患此病,未发现有种族差异,1型约占85%,2型约占15%。HAE是一种负担沉重的疾病,美国平均年治疗费为4.2万美元,轻、中、重度患者分别为1.4万、2.7万和9.6万美元[cxi]。尽管已经有多个产品获批用于HAE,但本品具有口服的优势,EVP预测本品在2026年的销售额可达3.26亿美元。
49. Klisyri(tirbanibulin)12月14日,FDA批准了Athenex 公司的tirbanibulin,用于面部或头皮的光化性角化病(AK)的局部治疗。Klisyri是一种软膏剂,含有1%的微管蛋白抑制剂tirbanibulin。FDA批准tirbanibulin上市,是基于两项随机、双盲、安慰剂对照的临床试验(NCT03285477和NCT03285490)的结果,702名面部或头皮有AK病斑的患者被1:1随机分为两组,分别每天一次,连续5天接受本品(n=353)和安慰剂(空白基质,n=349)治疗。试验的主要终点为治疗后57天,AK病斑完全清除的患者比例。试验一的结果显示,本品治疗组病斑被完全清除的患者比例为44%,而空白介质治疗组病斑被完全清除的患者比例为5%。试验二的结果与试验一类似,本品治疗组病斑被完全清除的患者比例为54%,而空白介质治疗组病斑被完全清除的患者比例为13%[cxii]。AK是一种慢性疾病,主要分布在阳光直射的皮肤区域,据估计每年有0.025%至16%的AK病斑最终转化为浸润性鳞状细胞癌。目前尚无权威的AK发病率,据Mahizer Yaldiz报道,AK在54786受访者中的患病率为2.50%,患病情况与年龄正相关,80岁以上人群患病率高达14.57%[cxiii],EVP预测该产品在2026年的销售额可达1.33亿美元。
50. Margenza(margetuximab-cmkb)12月16日,FDA批准了Macro Genics公司的margetuximab,适应症是与化疗联用,治疗既往接受过两种及以上抗HER2方案治疗的转移性HER2阳性乳腺癌成年患者。Margetuximab是一种新型HER2抗体,FDA批准其上市是基于一项名为SOPHIA的关键性三期临床研究(NCT02492711)的结果。536名既往接受过曲妥珠单抗或ado-trastuzumab emtansine治疗(一名患者接受过帕妥珠单抗治疗)的患者,在化疗的基础上分别接受margetuximab(n=266)或曲妥珠单抗(n=270)治疗,结果显示,margetuximab相比曲妥珠单抗降低了24%的死亡风险,总缓解率分别为22%和16%,中位PFS 分别为5.8和4.9个月[cxiv],OS数据尚未成熟。HER2阳性患者约占乳腺癌的15%[cxv],尽管已有多个HER2阳性的乳腺癌疗法获批,但仍存在未满足的临床需求,另外,margetuximab还在开发胃癌、食道癌等适应症,EVP预测该产品在2026年的销售额为2.77亿美元。
51. Orgovyx(relugolix)12月18日,FDA批准了Myovant公司的relugolix,用于治疗晚期前列腺癌成人患者。Relugolix是一种口服非肽类促性腺激素释放激素(GnRH)受体阻断剂,由武田、ASKA和Myovant共同开发,此前已经在日本获批用于子宫肌瘤治疗。FDA批准Orgovyx上市是基于一项名为HERO (NCT03085095)的临床研究结果,这是一项针对一年以上去势治疗后复发的晚期前列腺癌患者设计的随机、开放标签的临床试验。934名受试者按2:1的比例随机接受本品或亮丙瑞林微球治疗48周,结果显示,本品治疗组(n=622)29天至48周的持续去势(睾酮浓度<50ng/dL)率为96.7%,而亮丙瑞林微球组(n=308)为88.8%。Orgovyx治疗组在第8天就已经有91%的患者达到去势标准,而亮丙瑞林微球组为0%[cxvi]。试验结果表明,Orgovyx的疗效优于亮丙瑞林,而且起效更快,除了子宫肌瘤、前列腺癌,本品还在开发子宫内膜异位症引起的疼痛、痛经等适应症,EVP预测该产品在2026年的销售额为1.74亿美元。。
52. Ebanga(Ansuvimab-zykl)
2020年12月21日,FDA批准了Ridgeback Biotherap公司开发的ansuvimab,用于治疗成人和儿童的Zaire埃博拉病毒感染。Zaire埃博拉病毒是4种埃博拉病毒的一种,在此之前,FDA曾批准了Inmazeb对抗该病毒。Ebanga是一种糖蛋白抑制剂,可阻断病毒与细胞受体的结合,阻止其进入细胞。FDA批准该产品是基于一项2018年-2019年在刚果民主共和国(DRC)的埃博拉疫情期间,美国国立卫生研究院(NIH)和刚果民主共和国国家生物医学研究所共同牵头开展的一项名为“PALM”的临床研究。在该试验中,174名确诊患者(120名成人和54名儿童)接受了Ebanga治疗,168名患者(135名成人和33名儿童)则接受了ZMapp治疗(控制组),主要治疗终点是28天死亡率。结果显示,接受Ebanga治疗的174例患者中,28天死亡率为35.1%,而接受对照的168例患者中,28天死亡率达49.4%[cxvii]。
53. Gemtesa(vibegron)12月23日,FDA批准了Urovant Sciences的vibegron,用于伴有尿频、尿急、尿失禁的膀胱过度活动症(OAB)成人患者治疗。Vibegron是一种肾上腺素β3受体激动剂,作用机制与米拉贝隆相同,最早在2018年于日本获批上市[cxviii]。FDA批准vibegron上市主要是基于一项随机、双盲、安慰剂对照的三期临床试验(NCT03492281)的结果,在该试验中,1515名患者按5:5:4的比例随机接受本品、安慰剂、活性对照药治疗12周,入组的受试者均伴有三个月及以上的OAB症状,包括平均每日排尿至少8次和尿失禁至少1次,或平均每日排尿至少8次和尿急促至少3次。主要终点为12周时的每日平均排尿次数和尿失禁次数相比基线的改善。试验结果显示,接受本品治疗的526名患者,日均排尿次数、尿失禁和尿急次数分别相比基线下降了1.8次、2.0次和2.7次,而接受安慰剂治疗的520名患者,日均排尿、尿失禁和尿急次数分别相比基线下降了1.3次、1.4次和2.0次,均达统计学显著差异[cxix]。OAB是一种较为常见的疾病,流行病学数据显示,40岁以上东亚人群的总体患病率高达20.8%,其中女性为22.1%,男性19.5%,患病情况与年龄存在正相关,从40-44岁年龄段的10.8%逐渐上升到60岁以上年龄段的27.9%[cxx],EVP预测该产品在2026年的销售额可达4.25亿美元。