

对于药品上市许可,国际普遍采用药品上市许可持有人(MAH)制度,由药品批准证明文件的持有人承担药品全生命周期的法律责任。本文围绕药品注册审评程序,为您讲述中美欧药品上市许可申请和审评流程的区别。
中国注册审评程序
新<药品注册管理办法>(以下简称新<办法>)第三十四至三十九条明确了三种申请药品上市许可的路径:1)完整的申报路径;2)经申请人评估无需或不能开展药物临床试验,符合豁免药物临床试验条件的,申请人可以直接提出药品上市许可申请的路径;3)非处方药可以直接提出上市许可申请的路径。
对于完整的申报路径,根据新<办法>规定,申请人在完成支持药品上市注册的药学、药理毒理学和药物临床试验等研究,确定质量标准,完成商业规模生产工艺验证,并做好接受药品注册核查检验的准备后,提出药品上市许可申请。国家药监局药审中心(以下简称药审中心)对申报资料进行形式审查,符合要求的,5个工作日内予以受理。药审中心应当组织药学、医学和其他技术人员进行审评。审评过程中基于风险启动药品注册核查、检验(申请人可将药品注册检验调整为受理前启动)。药审中心根据药品注册申报资料、核查结果、检验结果等,对药品的安全性、有效性和质量可控性等进行综合审评。综合审评结论通过的,批准药品上市,发给药品注册证书;综合审评结论不通过的,作出不予批准决定。标准审评审批时限为200个工作日,优先审评时限为130个工作日。
新<办法>将原来的审评、核查和检验由“串联”改成“并联”,依据产品创新程度和风险特点,实行基于风险的审评、核查和检验模式,提高审评审批工作效率。我国药品上市许可申请和标准审评流程图见图1。
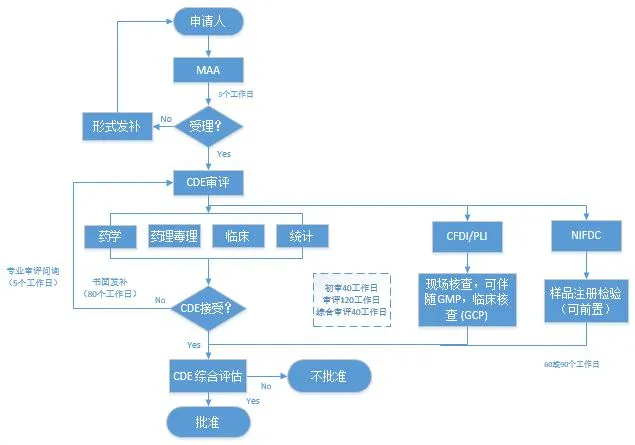
图1:中国药品注册审评流程图
美国注册审评程序
FDA将人用药品分为四大类,分别为处方药、非处方药、天然药物以及生物制品。处方药分为创新药(New Drug)和仿制药(Generics)两类,仿制药的上市采用<联邦食品、药品和化妆品法>(The Federal Food, Drug and Cosmetic Act,FD&C Act)505 (j)规定的ANDA批准程序。生物制品按照类别和监管属性,主要以新药申请(NDA)和生物制品许可(BLA)两类途径进行上市申请。生物制品作为特殊的药品,上市许可须满足<公共健康服务法>(Public Health Service Act,PHS Act)第351部分,以及FD&C Act第505部分“新药”的要求。2003年6月FDA把治疗用生物制品的审评、监管权由生物制品评价和研究中心(CBER)转移至FDA药品审评与研究中心(CDER),美国生物制品价格竞争与创新法案规定2020年3月23日之前按FD&C Act 505途径批准的生物制品,自动被归为按照PHS Act 351的BLA。
新药和生物制品上市许可申请的审评程序包括:申请的受理、新药技术审评、现场检查、通知审评结果、双方交流(申请前沟通会议、中期会议、审评结束会议和其他会议)等。1992年,根据<处方药用户付费法案>(Prescription Drug User Fee Act,PDUFA),FDA同意加快药物审评并且制定了标准审评(Standard Review)和优先审评(Priority Review)两种审评系统。2002年修订的PDUFA规定新药申请的标准评审应在10个月内完成。优先审评使评审时间缩短至6个月。美国FDA标准审评和优先审评时间表见图2。
FDA强制要求申请人以电子通用技术文档(eCTD)格式递交资料。
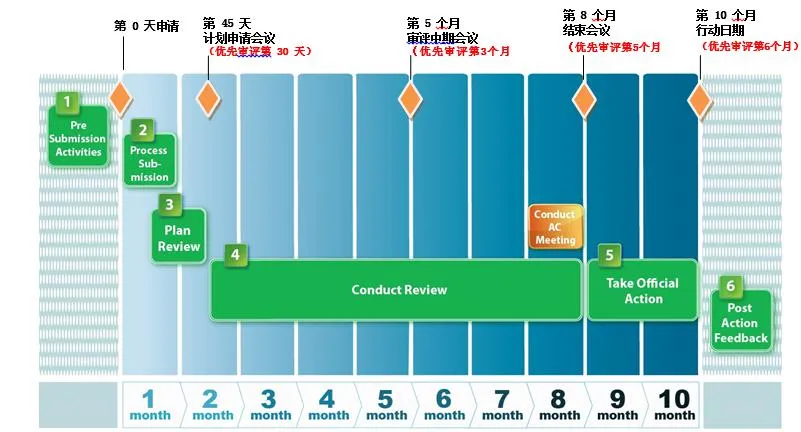
图2: FDA标准审评和优先审评时间表
注:图片来源于FDA CDER 21st Century Review Process
欧盟注册审评程序
药品在欧盟的上市申请(MAA)包含两种方式:国家授权方式和集中授权方式。其中,集中授权方式即“集中审批程序”(Centralized Procedure,CP),通过此程序获得上市许可的药品,可在所有欧盟成员国上市。国家授权方式对应三种注册程序,包括相互认可程序(Mutual Recognition Procedure,MRP)、分散审批程序(Decentralized Procedure,DCP)以及单一成员国审批程序(National Procedure,NP)。集中审批程序是新药和生物制品迅速进入整个欧盟市场最有效的途径。通过集中审批程序获得上市许可的药品,被强制性在欧盟内任意成员国的市场自由流通和销售。集中审批程序是欧盟对于药品管理的大趋势,EEC/726/2004法规明确规定:2005年11月20日起,适应症为艾滋病、肿瘤、神经退化疾病和糖尿病的药品必须按照CP申请;2008年5月20日起,适应症为病毒性疾病、自身免疫性疾病以及免疫相关疾病的药品必须按照CP申请。CP申请人应直接向欧洲药品管理局(EMA)提出药品上市许可的申请,EMA在对药品进行申报资料审评后,向欧盟委员会提出意见,欧盟委员会依据此意见决定是否颁发该产品在欧盟的药品证书。标准审评时限为277个日历日,加速审评为150个日历日。
欧盟药品集中审批程序注册和标准审评流程见图3。
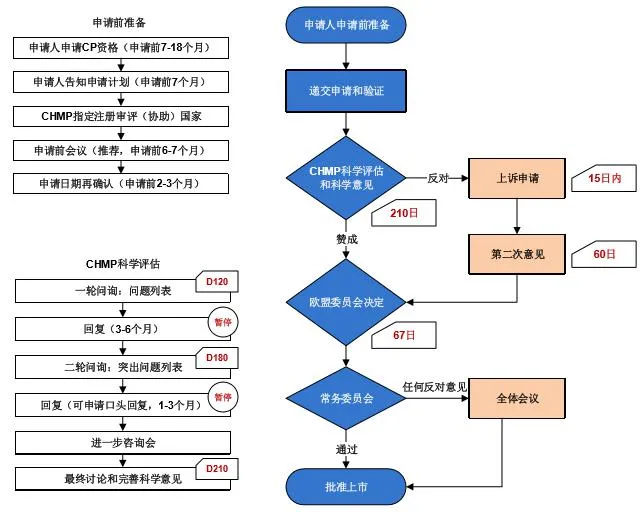
图3 欧盟药品集中审批程序注册审评流程图
中国、美国和欧盟上市许可注册审评对比分析
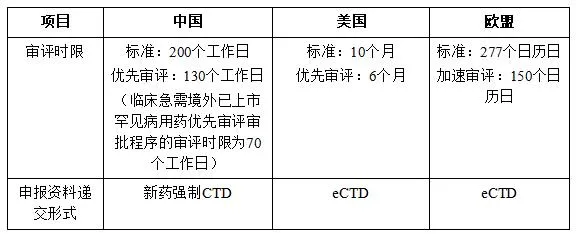
表1 中国、美国、欧盟审评时限对比表
审评时限和审评机构方面,新<办法>做到了与国际接轨,引入项目管理员(PM)与FDA的RPM角色相当;审评时限上突飞猛进,200个工作日与FDA的标准审评时限10个月以及欧盟的277天接近。另外,主要审评机构的职能和参与方式与FDA类似,只是我国的审评人员数量远远少于美国和欧盟。
审评程序方面,我国CDE审评由原来的“串联”(先审评、再核查、后检验)改为“并联”(审评、核查、检验同时进行),大大提高注册效率;同时,新<办法>解读中对审评做了更细节的规定,初审(40个工作日)、正式审评(120个工作日)、最后综合审评(40工作日),最终规定200个工作日的审评时限,和EMA的Primary(120个日历日)、Secondary(90个日历日)、Decision(67个日历日)的三个阶段分类相似,但是中欧在初审和正式审评阶段的侧重点各有不同。中国的初审与FDA 六个阶段中30天的预审评时长相当,希望未来可以通过初审将申报资料中存在的重大问题对申请人提出,这样进入正式审评后就无需面对重大缺陷或者大量补充数据,避免审评后期因重大发补而拖延时长。
根据新<办法>第一百零三条,申请人补充资料、核查后整改以及按要求核对生产工艺、质量标准和说明书等所占用的时间不计入审评时限。美国信息请求和学科审评函则不影响审评时限,根据申请人递交的内容是否属于重大增补及递交距离审评周期结束的时间,延长审评时限或暂缓审评。笔者认为,如果我国能在200工作日的审评阶段加入必要的主动沟通交流机制,则有利于申请中重大问题的解决,进一步提高审评效率。
内部控制方面,美国的PDUFA中明确规定了关于审评人员内部控制流程和时限要求。笔者认为,我国可以合理借鉴有关内容,加强对审评人员的内部管理。
申报资料方面,中美欧最大的差异在于模块一地域性文件,中国的要求相对复杂,需要提供的证明性文件较多;其余CTD章节框架基本相同。另外,欧美以eCTD格式递交为主,而中国目前主要以纸质格式递交,eCTD申报正在建设中。